A methodological article in the JBC
All scientific results must be made public. Anyone who develops a method in the course of his own research inevitably realizes that it could be of value to others, too. Erik Hrabovszky's group has developed a new RNA sequencing method, which has been accepted for publication in the Journal of Biological Chemistry, a true classic that has maintained its reputation and prestige for almost 120 years.
Nobel Prizes are not only awarded for great discoveries, for theories that open up new horizons, but also for the development of tools and methods that help or enable research. True, sometimes it demands a lot of luck and a long life. Ernst Ruska, who built the first electron microscope with Max Knoll in 1931, had to wait for 55 years before he could take the precious award in the company of Gerd Binnig and Heinrich Rohrer, who built the prize-winning scanning tunneling microscope five years earlier, in 1981. Moreover, Ruska was preceded by Aaron Klug too, who got his Nobel Prize in 1982 for the development of the crystallographic electron microscope and for studying the structure of nucleic acid-protein complexes.
We could cite some of the discoverers and developers of those important methods, which are now considered fundamental, however, we mention only the English Frederik Sanger. We have two very good reasons to do this. His two Nobel Prizes.
Up until 2022, Sanger was the only person to win two Nobel Prizes in Chemistry. (K. Barry Sharpless, one of the inventors of "click chemistry", was the other to win his second Nobel Prize in 2022.) This very modest researcher, who never overestimated himself or his achievements - and who, on reaching retirement age, had to tend to his garden from one day to the next, leaving his laboratory to younger people - worked very hard and never gave up between 1943 and 55, until he succeeded in determining the sequence of amino acids that make up the insulin molecule. This was a fine achievement in itself, a true world record, but the greatest good news was that his method could also be used to determine the primary structure of other proteins. He deservedly received his first Nobel Prize in 1958. By then, he was working on the big and fashionable research topic of his time, the structure of double-stranded DNA. Deciphering the sequence of nucleotides that make up DNA was a much more difficult and complex task, but his consistent, thoughtful work and perseverance paid off, and he was awarded another Nobel Prize in Chemistry in 1980 for his work on the structure of a bacteriophage DNA. (Half of the prize went to Paul Berg for the technology he devised to create the first recombinant DNA, and Walter Gilbert shared the other half for his own method of determining the structure of small DNAs relatively quickly.)
Another group of nucleic acids is RNA. While all the nucleated cells in our cells contain the same DNA, there are many types of RNA expressed, and their types depend on the cell type. This diversity should be taken literally! The so-called transcriptome, the complete set of RNA transcripts produced in a given cell or organism, is so large and complex that it is only worth studying using high-throughput methods. The discovery of PCR (polymerase chain reaction) for which a Nobel Prize in Chemistry was awarded to Kary B. Mullis in 1993, has played a key role in the emergence of a new generation of sequencing techniques. These techniques offer much greater precision and dynamic range than the previous methods.
The work of Erik Hrabovszky's group allows the transcriptomic characterization of neuronal cell types defined in terms of both neurochemistry and anatomical location. Their paper was published in the highly regarded Journal of Biological Chemistry (JBC), which, although not a journal specializing in neuroscience, has always sought to be a forum for related disciplines, alongside biological chemistry - biochemistry, thus helping to unravel the molecular and cellular basis of biological processes.
We spoke to Erik Hrabovszky, the group's leader, about their work.
- Congratulations on your publication with shared first authorship with Éva Rumpler and Balázs Göcz! What's worth knowing about it even as a non-expert?
- This is a communication describing in detail a methodology that can be used in a variety of applications, and in which a number of partial results obtained during the optimization of the method are also presented.
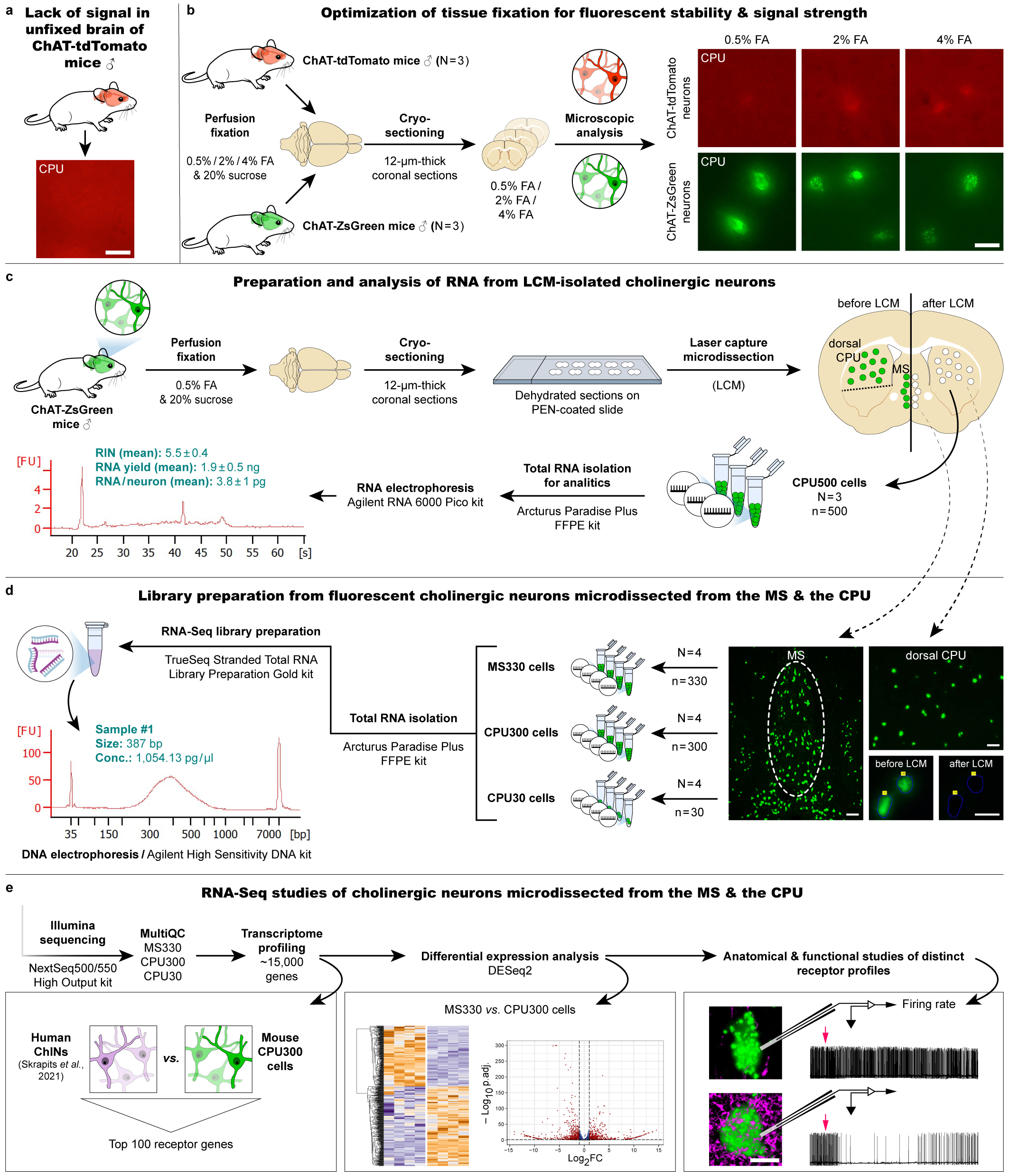
When we apply our "LCM-Seq" (laser capture microdissection - sequencing), neurons expressing the ZsGreen luminescent protein from genetically modified animals are collected by LCM from an arbitrarily selected brain region, RNA is isolated, a cDNA library is constructed, and a new generation of sequencing is performed to reveal the complete transcriptome of the neuron.
- Since the use of lasers for more than just eye surgery has been common practice for several decades, it is easy to believe that it is possible to excise and cleanly collect certain cell groups from specific areas under the microscope. A cDNA library is a transcript of mRNA isolated from the cells you collect, using a process called reverse transcription. But how much of an improvement is your Ti method compared to existing sequencing methods??
- Ours can also be classified as one of the many spatial transcriptomics methodologies that use very different strategies. The spatial information retention, the fact that the sample to be sequenced comes from precisely defined locations, is ensured by LCM-based cell isolation from histological sections.
- And what ensures that your results are reliable for each cell type?
- Highly sensitive detection of the cell-type-specific transcriptome was achieved by using hundreds of "pooled" (pooled together) neurons collected from the same site. This allows the identification of about 15,000 different transcripts, which exceeds the sensitivity of various single-cell RNA sequencing (scRNA-Seq) methods, which are usually capable of detecting 3-4,000 different RNAs per cell. Of course, these methods also have unique advantages.
- Thousands of times more RNA isolates are used. Why is this important?
- In addition to the high sensitivity, which allows the detection of rare transcripts, another very important advantage comes from the fact that sequencing starts from RNA isolates of nanogram scale, compared to the picogram scale of single-cell sequencing methods. The larger starting RNA amount also facilitates the reliable quantification of individual RNA molecules. An example of this is presented in the paper. Using transcriptomic comparisons of two cholinergic cell types as a model, we detected 2800 significant differences between cholinergic cell types in two brain regions, the medial septum, and striatum
.- The sensitivity and suitability of your methods for quantitative studies is truly impressive. What types of questions will it be used to answer and where is such a high level of reliability and accuracy needed?
- In principle, the transcriptome of any neuronal system in which the product of a fluorescent transgenic protein provides a signal of sufficient intensity for the microscopic identification of cells can be analyzed using this approach.
For an example of when such precision is required, I can cite our paper from last year (Göcz et al., PNAS), in which we compared the transcriptome of kisspeptin neurons from ovariectomized and then estrogen- or vehicle-treated mice. Here, the methodological precision mentioned above allowed the reliable identification of ~2300 estrogen-dependent genes. Systematic optimization of the method was performed in individual experiments of the present Methods paper.
- It already gave excellent results then, but you refined it further. Why was this necessary?
- Preservation of fluorescent signals requires tissue fixation with formalin, while for next-generation sequencing, unfixed tissues provide the best preservation of RNA for cDNA library construction. Tests were conducted to select the optimal fluorescent marker, the concentration of formalin, the ideal number of neurons to be isolated, and the technologies and commercially available kits for RNA isolation and library preparation. Balázs Göcz and Szabolcs Takács have also gained considerable bioinformatics skills during their work, which will facilitate future transcriptomics projects based on similar principles in our laboratory.
- Are you involved in further development of the methodology and, if so, what is the direction of development?
- In the LCM-Seq study of fluorescent neurons described in the JBC article, we used a genetically modified mouse model. This is a limitation that has recently been overcome. We can now also study neurons identified by immunohistochemistry in formalin-fixed tissues!
- Then you can also test older samples! This is great. And what will be the most exciting part?
- The most exciting area of research will of course be to unravel the transcriptome of specific human nerve cell types from post-mortem histology samples. We have already been successful in this endeavor for several peptidergic cell types.
Éva Rumpler, Balázs Göcz, Katalin Skrapits, Miklós Sárvári, Szabolcs Takács, Imre Farkas, Szilárd Póliska, Márton Papp, Norbert Solymosi, Erik Hrabovszky: Development of a versatile LCM-Seq method for spatial transcriptomics of fluorescently-tagged cholinergic neuron populations.